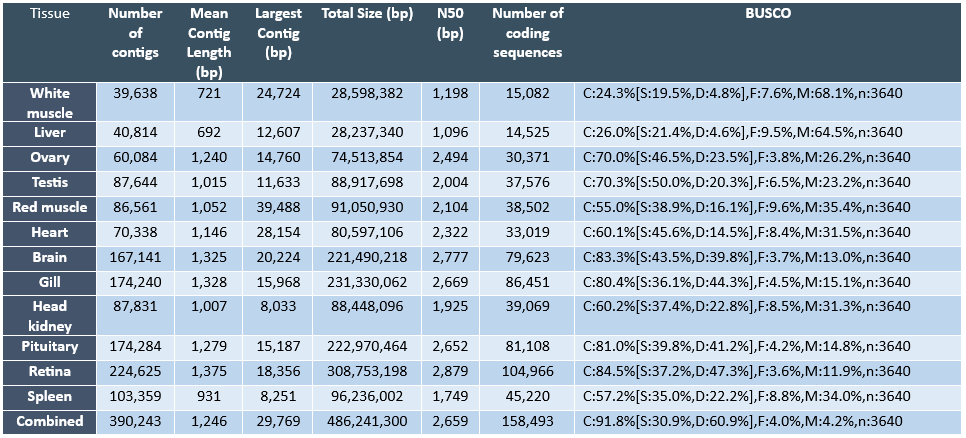
The ability to monitor changes in biological processes over time and space is particularly important given that these processes are potentially impacted by environmental and anthropological influences. Therefore, there is a great need to identify and develop novel indicators of biological changes in the Pacific halibut population to evaluate the immediate response of the population, as well as to predict biological changes in the future. In this regard, research approaches investigating the transcriptome (i.e. the collection of transcripts from expressed genes that in great part will be translated into proteins) are enormously useful to identify potential biological markers for a variety of reasons. First, given that the transcriptome is a reflection of the activation state of the genome (i.e. through the transcription or expression of the genome), transcriptomic assessment provides an opportunity to evaluate physiological responses of organisms to external influences at a molecular level. It is well known that gene expression changes represent some of the first and most important responses driving adaptive changes in organisms. Second, although not all genes present in a vertebrate genome (i.e. estimated to contain approximately 20,000 to 30,000 genes) are expressed at all times nor in all cells, the number of genes that are expressed at any given time or tissue under specific physiological conditions is very high (e.g. in the order of thousands) and, therefore, transcriptomic approaches can identify large numbers of potential marker genes for specific biological processes or tissues. Finally, currently available RNA sequencing (RNA-seq) technologies have become increasingly powerful and economical to allow an unprecedented view of the transcriptome of any species (Ozsolak and Milos, 2011) and this is particularly relevant for species without a sequenced genome, such as the Pacific halibut. Similar technical approaches have been adopted to generate genomic and transcriptomic resources in other flatfish species (Benzeki et al., 2014; Ribas et al., 2013). Therefore, IPHC has embarked on an initial characterization of the transcriptome of the Pacific halibut by conducting RNA-seq on twelve different tissues. The sequence data from each tissue was assembled separately per tissue and a combined assembly (i.e. combining all sequence reads from all tissues) has also been generated. Through this approach we have identified tissue specific molecular markers that can be used to monitor growth, reproduction, performance and condition in this species.
References
Benzekri H, Cousin X, Armesto P, Rovira M, Crespo D, Merlo MA, Mazurais D, Bautista R, Guerrero-Fernández D, Ponce M, Infante C, Zambonino JL, Nidelet S, Gut M, Rebordinos L, Planas JV, Begóut ML, Claros MG, Manchado M. 2014. De novo assembly, characterization and functional annotation of Senegalese sole (Solea senegalensis) and common sole (Solea solea) transcriptomes. Integration in a database and design of a microarray. BMC Genomics 15:952.
Ozsolak, F., Milos, P.M. 2011. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12:87-98.
Ribas L, Gómez-Pardo B, Fernández C, Alvarez-Dios JA, Gómez-Tato A., Quiroga MI, Planas JV, Sitjà-Bobadilla A, Martínez P, Piferrer F. 2013. A combined strategy involving Sanger and 454 pyrosequencing increases genomic resources to aid in the management of reproduction, disease control and genetic selection in the turbot (Scophtalmus maximus). BMC Genomics 14:180.